
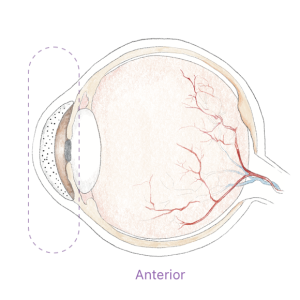
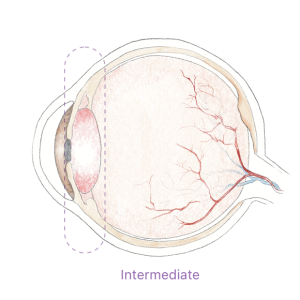
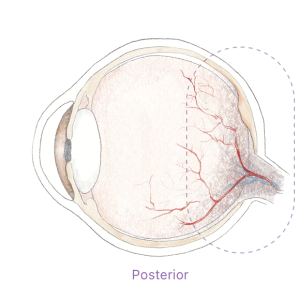


A rare, potentially chronic and blinding, inflammatory disease of the eye.
Uveitis can be classified as infectious (resulting from bacteria or virus) or non-infectious (autoimmune, idiopathic cause). Uveitis can occur in one or both eyes of a patient, in different segments of the eye
Uveitis can be anterior, intermediate, or posterior. The inflammation can be present in both eyes at the same time, affect one eye, or alternate from one eye to the other.
Patients in the U.S.
with different types of uveitis
Patients with uveitis may experience pain, light sensitivity/photophobia, eye redness, decreased vision, blurred vision, or “floaters”.
If not treated properly and promptly, uveitic flare-ups may lead to blindness.
of blindness in the U.S.
is due to uveitis
The most common form of uveitis
The cause is often unknown and is said to be autoimmune. In these cases, an inflammatory response in the eye arises even without a well-defined stimulus. The ocular inflammation is initiated by inflammatory macrophages that secret inflammatory cytokines. This leads to a cascade of events that eventually ends up in immune cells entering the anterior chamber of the eye, which is otherwise an ‘immune privileged’ site.
The main hallmark of non-infectious anterior uveitis is the presence of immune cells floating inside the anterior chamber of the eye, and the severity of the uveitis can be determined by the quantity of cells present, seen in a slit lamp examination.
With available treatment options, visual loss occurs in about two-thirds of uveitis patients, with 22% of patients meeting the criteria for legal blindness at some point of their follow-up.
Existing treatments often start with topical steroids, followed by systemic immunosuppressants (such as biologics, T-cell inhibitors, alkylating agents etc).
Steroid eyedrops are potent at resolving inflammation, but prolonged use entails severe and blinding side effects (i.e., glaucoma, intraocular pressure elevation, formation of cataracts etc.).
Immunosuppressants are prescribed to provide a long-term reduction of the number of attacks or prevent them altogether.
Their use requires more clinic visits to monitor their effect and avoid side effects including infections, diabetes, and in some cases cancer. As a result of serious safety concerns, many systemic immunosuppressive drugs are not approved for pediatric use.
Recurrent exacerbations of uveitis can lead to cumulative damage and cause tissue destruction and scarring, and a variety of complications, such as retinal swelling (macular edema), retina scarring, glaucoma, cataracts, optic nerve damage, retinal detachment, low eye pressure (hypotony) and eventually in some cases, permanent vision loss.
immunosuppressive treatment requires 2-12 weeks to take effect. As a result, topical steroids still remain the only available treatment for a new flare-up or active disease.

Tarsier Pharma is committed to help patients save their eyesight and see their future.
Served as a public service.
© 2023 Tarsier® Pharma
For more information email us:
info@tarsierpharma.com
Zichron Yaacov, Israel
We at Tarsier Pharma Ltd. (“Tarsier Pharma”, “we”, “us” or “our”) respect your privacy. This Privacy Policy (“Policy”) explains the accepted privacy practices for our online services at our website (“uveiticlgaucoma.com”). It also describes the rights and options available to you with respect to your personal information.
The Website provides content and information about Tarsier Pharma and its research and development of a novel platform for treatment of ocular inflammatory diseases.
You do not have a legal duty to provide the information and providing it is subject to your unconditional consent. Proving the service of online inquiries on our Website will not be available to those choosing not to provide the information requested.
We use your Information in order to contact you about Inquiries you made and/or to operate the Website.
We may use your Information outlined above for the following purposes:
Operating the Website and providing its features and functionalities;
Contact you about inquiries you made;
Send you email communications concerning our products, programs, developments and other related information. We will send you email communication subject to your explicit consent;
You may ‘opt-out’ of using your data for promotional communications at any time by following the “unsubscribe” link located at the bottom of each message. By doing so, Tarsier Pharma will only delete the Information which is required to contact you for promotional communications, while the rest of the Information you submitted to us which is necessary in order to provide you with the Website’s services will continue to be processed.
Improve and enhance the services and content offered on the Website.
We share your Information mainly with our contractors and service providers, strictly for the purpose of helping us with the internal operation of the Website.
We will not share your Information with third parties, except in the events listed below or when you provide your explicit and informed consent:
We will share your Information with our service providers (such as our hosting providers) who assist us with the internal operations of our Website and our services.
These companies are authorized to use your Information only as necessary to provide these services to us and not for their own promotional purposes. We do not sell your Information to third parties
If you have breached any agreement you have with us (including this Policy), abused your rights to use the Websites, or violated any applicable law, your Information may be shared with the competent authorities, law enforcement and third parties (such as legal counsels and advisors), for the purpose of handling of the violation or breach;
If we are required to disclose your Information by a judicial, governmental or regulatory authority;
We will share your Information with members of our family group of companies, who help us process the data for the purpose set out above;
If the operation of the Websites is organized within a different framework, or through another legal structure or entity (e.g. due to a merger or acquisition), we will share your Information to enable the structural change.
We will store your data for as long as we deem necessary for the purposes detailed in this Policy
We retain personal data for as long as necessary to fulfil the purposes we collected it for (as detailed above), including for the purposes of satisfying any legal, accounting, or reporting requirements. We, at Tarsier Pharma, always consider the type of information collected, the amount of information, how sensitive it might be. Accordingly, we determine the appropriate retention period.
We may also anonymize your Information so that it can no longer be associated with you, in which case we will use such Information without further notice to you.
We implement measures to reduce the risks of damage and unauthorized access or use of information, including when data is transferred outside the E.U.
Security Measures. We implement measures to reduce the risks of damage, loss of Information and unauthorized access or use of Information. We also request our affiliates to implement such measures in order to secure the Information we provide them. However, although efforts are made to secure your Information, we cannot guarantee its absolute protection.
Cross-Border Data Transfers. We are based in Israel. Information we collect from you will be processed in Israel, which is recognized by the European Commission as having adequate protection for personal data. Certain information may be stored externally via cloud services. We will only transfer your personal information to a country or company which has been deemed to have an adequate level of data protection by the EU commission, or under other adequate safeguards determined under the applicable law.
Controller. Tarsier Pharma Ltd. is the data controller for the purposes of the personal data we collect via the Website and for the performance of the services offered through the Website.
The legal basis for processing and collecting Inquiry Information is its necessity for the provision of feedback, comment or service in response to your inquiry;
The legal basis for processing and collecting Analytical Information is our legitimate interests in operating our website, ongoing management of our business and business development;
The legal basis for collecting and processing your information for Promotional Communications is your explicit consent;
Right to Access. You have a right to access your personal data that we process and receive a copy of it.
Right to Rectification. You have the right to ask us to rectify inaccurate personal data concerning you and to have incomplete personal data completed.
Right to Data Portability. You have a right to receive the personal data that you provided to us, in a structured, commonly used and machine-readable format. You have the right to transmit this data to a third party. Where technically feasible, you have the right that your personal data be transmitted directly from us to a third party you designated.
Right to Withdraw Consent. You have the right to withdraw your consent for processing your personal data at any time. If you do that, we will not collect any further personal data, but we will further process the data we already collected for reasons described in this Policy. Withdrawing your consent will not affect the lawfulness of data processing we carried out based on your consent before such withdrawal.
Right to Object. If you previously agreed that your personal data may be used for other purposes other than registering and/or placing an order, you may have a right to object to the use of your personal data for such additional purposes.
Right to Restrict. You have the right to restrict processing of your personal data (except for storing it) if you contest the accuracy of your personal data, for a period enabling us to verify its accuracy; if you believe that the processing is unlawful and you opposes the erasure of the personal data and requests instead to restrict its use; if we no longer need the personal data for the purposes outlined in this Policy, but they are required by you to establish, exercise or defence relating to legal claims, or if you object to processing, pending the verification whether our legitimate grounds for processing override yours.
Right to be Forgotten. Under certain circumstances, such as when you withdraw your consent, you have the right to ask us to erase your personal data. However, we may still process your personal data if it is necessary to comply with a legal obligation we are subject to under laws in EU Member States.
If you believe your right have been infringed, you can lodge a complaint with a supervisory authority operating under the GDPR. For a list of supervisory authorities in the EU, click here.
Representation for data subjects in the EU
We value your privacy and your rights as a data subject and have therefore appointed Prighter Group with its local partners as our privacy representative and your point of contact.
Prighter gives you an easy way to exercise your privacy-related rights (e.g. requests to access or erase personal data). If you want to contact us via our representative Prighter or make use of your data subject rights, please visit the following website. https://prighter.com/q/11999524960
From time to time, we may change this Policy. We will provide you notice of such changes through the Website interface.
You may contact us via our email at info@tarsierpharma.com, We will do our best to resolve your issue promptly.
Last update: April, 2022